VQE
In Divi, we offer two different VQE modes. The first one is a standard single-instance ground-state energy estimation, and the latter is the hyperparameter sweep mode. We will provide examples to demonstrate both modes in this section.
Vanilla VQE
For our VQE implementation, we integrate tightly with PennyLane’s qchem module. The VQE constructor accepts a Molecule object (from PennyLane) or a pre-built hamiltonian operator. When using a Molecule, the molecular Hamiltonian is generated automatically.
The constructor also accepts an ansatz (from Divi’s ansatz library), an optimizer, and the maximum number of optimization iterations. An example of how to initialize a VQE object is shown below:
import time
import numpy as np
from pennylane import qchem
from divi.backends import MaestroSimulator
from divi.qprog import VQE, HartreeFockAnsatz
from divi.qprog.optimizers import ScipyOptimizer, ScipyMethod
molecule = qchem.Molecule(
symbols=["H", "H"],
coordinates=np.array([(0.0, 0.0, 0.0), (0.0, 0.0, 0.5)]),
unit="angstrom",
)
vqe_problem = VQE(
molecule=molecule,
ansatz=HartreeFockAnsatz(),
optimizer=ScipyOptimizer(ScipyMethod.L_BFGS_B),
max_iterations=3,
backend=MaestroSimulator(),
)
vqe_problem.run()
energies = vqe_problem.losses[-1]
print(f"Minimum Energy Achieved: {min(energies.values()):.4f}")
print(f"Total circuits: {vqe_problem.total_circuit_count}")In the example above, we attempt to compute the ground state energy of a hydrogen molecule (H₂). To extract the energy at the
end of the optimization step, we simply access the last item of the losses class variable, which stores the
losses of each iteration in the form of a dictionary mapping a parameter’s ID to its actual values.
For L-BFGS-B, an iteration uses one set of parameters, and so the min(energies.values())
bit you see in the example is a bit redundant. If we were to use Monte-Carlo sampling, we would have as many losses
as the sample points, and so the use of the min function becomes more salient.
Ansatze
Divi provides several ansatz choices for VQE:
| Ansatz | Description |
|---|---|
HartreeFockAnsatz |
Chemistry-inspired Hartree-Fock initial state |
UCCSDAnsatz |
Unitary Coupled Cluster Singles and Doubles |
HardwareEfficientAnsatz |
Hardware-native entangling layers |
GenericLayerAnsatz |
Custom gate sequences repeated in layers |
You can also provide a pre-built hamiltonian (PennyLane operator) directly instead of a Molecule object:
vqe_problem = VQE(
hamiltonian=my_hamiltonian,
ansatz=UCCSDAnsatz(),
n_electrons=2,
optimizer=ScipyOptimizer(ScipyMethod.COBYLA),
max_iterations=10,
backend=MaestroSimulator(),
)VQE Hyperparameter Sweep
By sweeping over physical parameters like bond length and varying the ansatz, this mode enables large-scale quantum chemistry simulations — efficiently distributing the workload across cloud or hybrid backends.
This mode is particularly useful for the study molecular behavior and reaction dynamics. It also allows one to compare ansatz performance and optimizer robustness. All through a single class!
The example below demonstrates how to use Divi’s VQEHyperparameterSweep class to
run a parallelized VQE simulation across multiple bond lengths and ansatz types for a hydrogen molecule (H₂).
import numpy as np
from divi.qprog import VQEHyperparameterSweep, HartreeFockAnsatz, UCCSDAnsatz
from divi.qprog.optimizers import MonteCarloOptimizer
from divi.backends import QoroService
q_service = QoroService()
vqe_problem = VQEHyperparameterSweep(
symbols=["H", "H"],
coordinate_structure=[(0, 0, 0), (0, 0, 1)],
bond_lengths=list(np.linspace(0.1, 2.7, 15)),
ansatze=[HartreeFockAnsatz(), UCCSDAnsatz()],
max_iterations=1,
optimizer=MonteCarloOptimizer(),
backend=q_service,
)
vqe_problem.create_programs()
vqe_problem.run()
vqe_problem.aggregate_results()
print(f"Total circuits: {vqe_problem.total_circuit_count}")
print(f"Simulation time: {vqe_problem.total_run_time}")
vqe_problem.visualize_results()What’s Happening?
| Step | Description |
|---|---|
VQEHyperparameterSweep(...) |
Initializes a batch of VQE programs over a range of bond lengths and ansatz strategies. |
symbols=["H", "H"] |
Defines a hydrogen molecule with two atoms. |
bond_lengths=... |
Sweeps bond distances from 0.1 to 2.7 Å in 15 steps. |
ansatze=[...] |
Runs two different quantum circuit models for comparison. |
create_programs() |
Constructs all circuits for each (bond length, ansatz) pair. |
run() |
Executes all VQE circuits — possibly in parallel. |
aggregate_results() |
Collects and merges the final energy values for plotting. |
visualize_results() |
Displays a graph of energy vs. bond length for each ansatz. |
Visualization
Divi comes built with visualization tools that allows the user to compare the approaches. The above example produces this plot for example. This is an ongoing effort, the goal is to provide dashboards for better visualization and a more in-depth comparison.
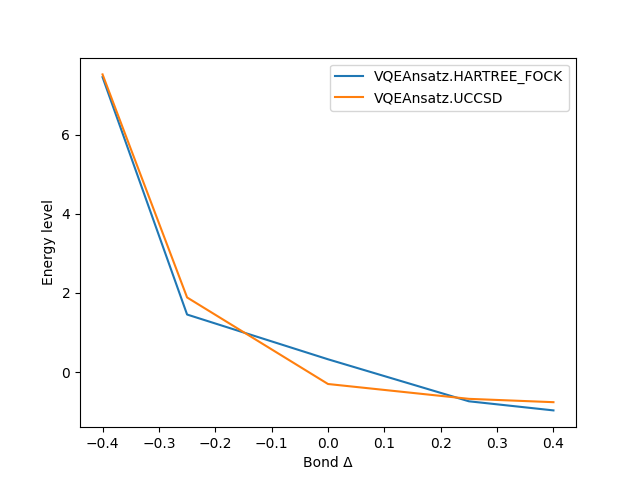
The result of running parallelized VQE
Why Parallelize VQE?
- VQE is an iterative algorithm requiring multiple circuit evaluations per step.
- Sweeping over bond lengths and ansatze creates hundreds of circuits.
- Parallelizing execution reduces total compute time and helps saturate available QPU/GPU/CPU resources.